mRIN for direct assessment of genome-wide and gene-specific mRNA integrity from large-scale RNA-sequencing data
Abstract
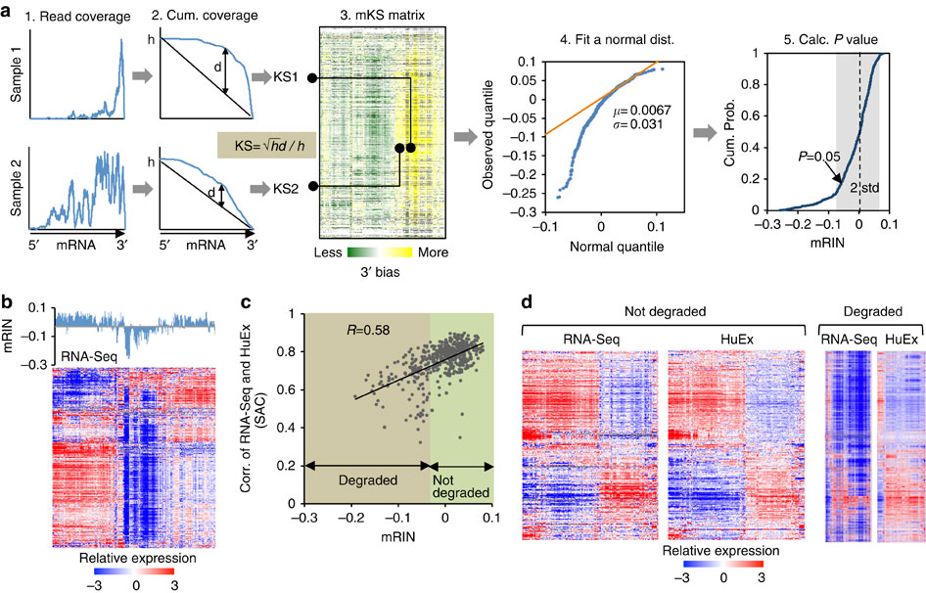
The volume of RNA-Seq data sets in public repositories has been expanding exponentially, providing unprecedented opportunities to study gene expression regulation. Because degraded RNA samples, such as those collected from post-mortem tissues, can result in distinct expression profiles with potential biases, a particularly important step in mining these data is quality control. Here we develop a method named mRIN to directly assess mRNA integrity from RNA-Seq data at the sample and individual gene level. We systematically analyse large-scale RNA-Seq data sets of the human brain transcriptome generated by different consortia. Our analysis demonstrates that 3′ bias resulting from partial RNA fragmentation in post-mortem tissues has a marked impact on global expression profiles, and that mRIN effectively identifies samples with different levels of mRNA degradation. Unexpectedly, this process has a reproducible and gene-specific component, and transcripts with different stabilities are associated with distinct functions and structural features reminiscent of mRNA decay in living cells.